
Mapping the off-target effects of every FDA-approved drug in existence (EvE Bio)
6.2k words, 29 minutes reading time


Note: Thank you to Bill Busa, CEO and co-founder of EvE Bio, for an extremely helpful discussion while working on this essay.
This essay is long, and I recognize that many people don’t necessarily care about the details. The real headline point you need to be aware of is this dataset, which was produced by EvE Bio underneath a CC-NA license, and is a comprehensive mapping of the interactions between a significant fraction of clinically important human cellular receptors and 1,600~ FDA-approved drugs. I strongly believe that this data is really, really useful, and more people should be aware it exists.
If you’d like to understand why I think it is useful, and what the dataset exactly contains, read on!
Introduction
If you were to be a fly on the wall during the 1-6 years of preclinical drug discovery research within a pharmaceutical company, one observation you may walk away with is that, while the work is certainly complicated, it is also frighteningly limited in scope. What you’ll learn is that drugs are made by corporations that are optimizing for one primary thing, and one thing only: work. ‘Working’ is obviously contextual, but it is a simple concept no matter the situation: reduce a worrying biomarker, improve mood, lengthen lifespan and so on and so on. What does this discovery process ignore? Simply put: everything else a drug could do beyond that.
Yes, that’s a roundabout way of describing ‘off-target effects’ — defined as the action of a drug at a gene product other than the gene product it was intended to affect — but I think it’s a helpful intuition pump. Viewing the drug discovery process as ‘not paying attention to anything that is unrelated to the drug working’ is useful in that it contextualizes the situation we’re in. Drugs are meant to make money, and money is derived from drugs working. To spend time on understanding what else a particular drug does beyond It Working for its intended task is time lost and money lost.
One unfamiliar with the drug discovery process may find this bizarre; why wouldn’t the well meaning scientists in charge of developing drugs try to deeply understand how it interacts with the body? On the other hand, those deeply in the medical field would find this thesis so obvious that stating it is unnecessary; of course a pharmaceutical company would limit their scope of understanding a drug to things that lie between it working and not working. There’s only so much time and resources to go around. Priorities!
Of course, if an off-target effect comes between the drug and It Working, then certainly resources will be allocated to deal with it. But beyond that, mapping everything else a clinical-stage drug does — every receptor it unintentionally binds, every pathway it nudges sideways, every gene it perturbs slightly — is deemed so high effort and so low ROI, that it is relegated to hoping an academic will study it. Only if post-marketing surveillance turns up something worrying shall further exploration occur. Because, again, a deep understanding of what exactly an exogenous chemical is doing inside a body is not the point of the drug discovery process. Working is the point!
With that background context, I am ready to present three claims I’m going to make in this essay and spend the remaining sections trying to prove:
Understanding off-target effects is really useful.
Learning about off-target effects at scale is possible.
No for-profit institution has a strong incentive to do this work.
For the moment, let’s accept that these three are indeed true, and we can put our skeptic hat back on at the end of this section.
The subject of today’s essay is EvE Bio, and why I think they are doing something incredible.
EvE is a bit unlike the typical startups I write about, because they aren’t really a startup. They are a FRO, or Focused Research Organization. Many reading this blog are likely already familiar with this recent renaissance of strange scientific organizations (something I’ve written about in the past) and already understand this acronym, but to those who don’t, this Venn diagram is quite instructive:
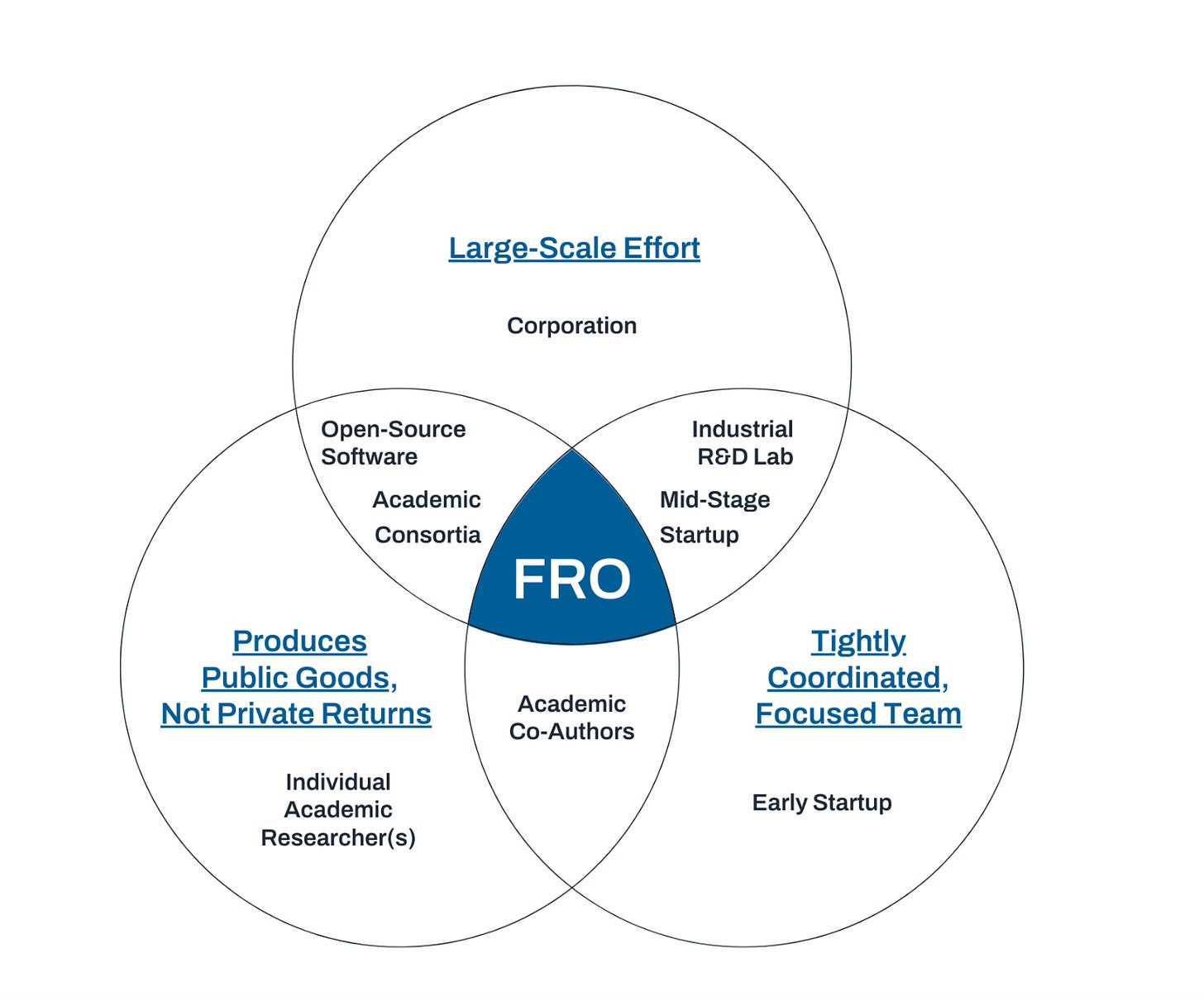
Entire essays can (and have been!) written about the intricacies of FRO’s, but this essay will ignore much of their organizational structure, since it isn’t super relevant to what EvE is doing.
So what is EvE doing? EvE Bio is a scientific non-profit that has a clear, singular mission: map the off-target effects of every FDA-approved drug in existence and share the data. The data will be released underneath a non-commercial, creative commons license — free to use by academics, and available for licensing for commercial entities. Once they accomplish this task, they close up shop or spin off into their own thing. And if they don’t do it within 5-6 years, the same end result still happens. They do have some future plans that may come into the picture with time, which I’ll cover at the end, but the bolded bit is their primary thesis!
So why are they doing this? How will they do it? And why hasn’t anyone else done it yet?
Why is understanding off-target effects important?
There is a lazy answer that could be given here: "because we want to know if potential side effects of a drug exist". This is partially correct, but I think it pays to be more specific. On EvE’s website, they list six reasons why off-target effects are worth studying:
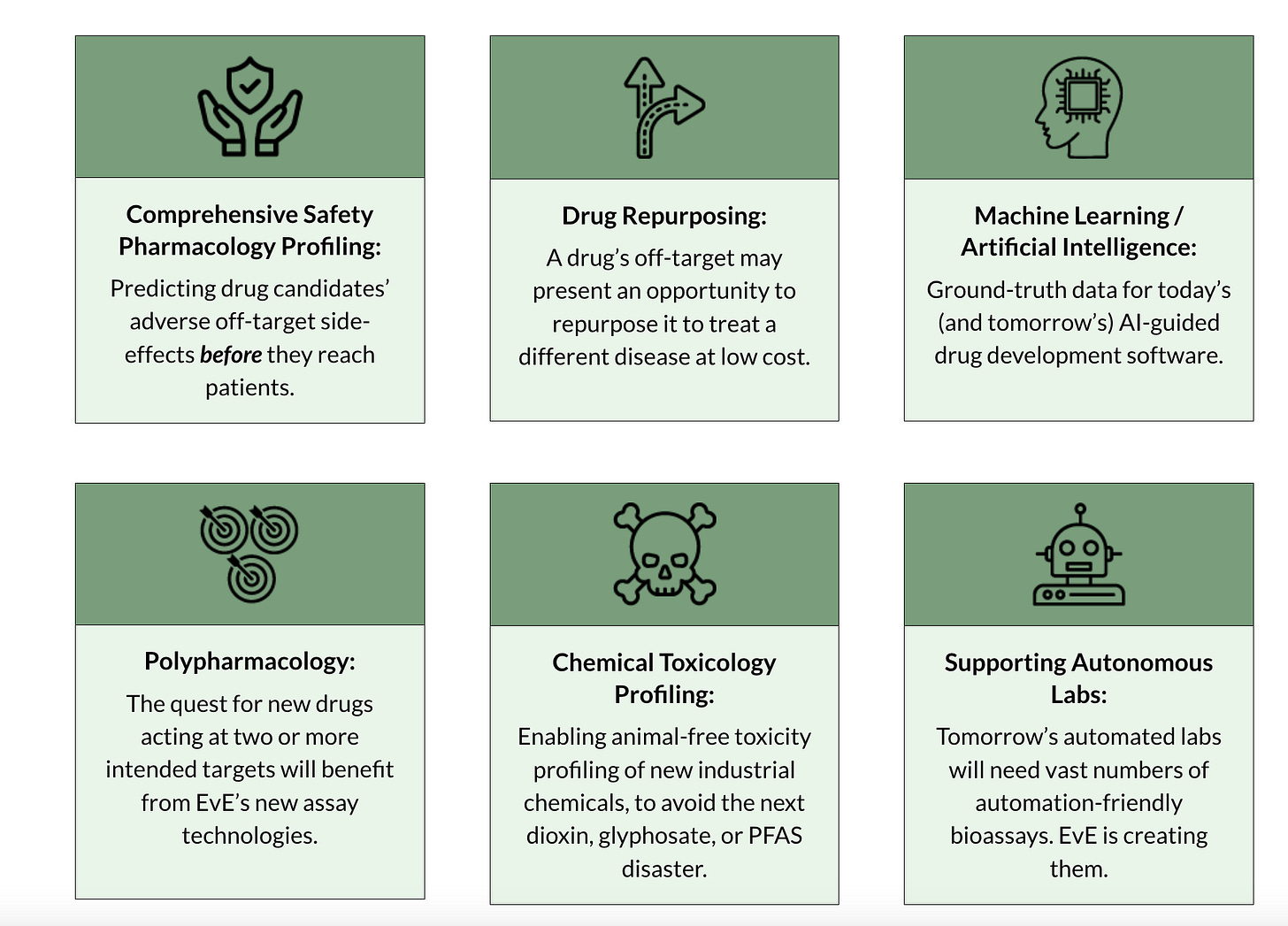
Now, fairly, some of these are at least a little fluffy. Is the institution doing off-target mapping really going to be the ones developing the autonomous lab assays of the future? Maybe! But it feels like a third-order, fourth-order, or even further consequence of their main mission. Bill did mention to me that there are already promising results in that direction, such as better reporting cell lines, but still. I think it’s generally good to limit ones assessment of an institution based on what their first-order impact will be, and, there, I think there will be three distinct areas that EvE will service: drug repurposing, validation for machine-learning models, and to a weaker degree, polypharmacology.
What about industrial chemical profiling and pharmacology profiling? I think EvE will certainly be important there, but it’s a bit fuzzier. Industrial chemical profiling may occur in the future but isn’t part of the current cohort of FDA-approved drugs that EvE is focusing on, and there’s a similar problem for pharmacology profiling as there is for ML-for-toxicity (which I have written about before as being a challenging proposition).
But even if we take my somewhat pessimistic stance that only three of these six things are genuinely tractable in the short term, those areas alone are extremely valuable. Let’s go over them.
Drug repurposing
I think it is under-appreciated just how rich the cohort of FDA-approved drugs that are out there. Consider the fact that basically all drugs start off with singular indications, meant to cure, alleviate, or address one thing. Yet, 30%~ of FDA-approved drugs gain a new post-approval indication, based on a study of the 197 drugs approved by the FDA from 1997-2020. Funnily enough, the same paper that came up with that 30% number almost treats it as a matter of disappointment, given that 38% of all prescriptions written in the US are off-label! This implies that there are, potentially, hundreds of drugs that are already being used beyond their original scope, just without the formal validation or regulatory blessing. Which, in turn, implies that we’re sitting on a vast, under-explored landscape of therapeutic potential, one that clinicians are already intuitively poking into, but which the formal system has barely begun to chart.
Now, I think some caution is warranted. This 38% number does vary from paper to paper, one other study claims off-label prescriptions are as low as 25%. If we’re being even more fair, it’s questionable exactly how proven-out these off-label indications are. One 2006 study claims that of the 21% of off-label prescriptions they found, 73% of them had little-to-no scientific support. Hard to tell whether this is because there simply are no studies, or because the off-label usage was actively disproved!
Consider gabapentin, one of the most egregious cases of off-label drug prescriptions. Typically, most people view gabapentin as the nerve injury drug, right? But it, in fact, was not originally approved for that, only for seizures! Yet, 95% of its prescriptions usage are for pain; nerve pain, low-back pain, post-operative pain, and so on. But while the gabapentin is indeed effective for some specific types of nerve pain (diabetic neuropathy), it is ineffective for many other types (e.g sciatica), as confirmed by follow-up studies by Pfizer.
Yet, prescriptions for these ineffective off-label usages continue.
But even if the true rate of valid, effective off-label use is lower than we’d like to imagine, the value of actually stumbling across a chance to repurpose a drug is high enough as to almost certainly still be worth it! Why? New chemical entities must follow the typical clinical phase progression timeline, whereas any repurposed drugs can skip preclinical, phase 1, and (sometimes) phase 2 trials as a result of their already-collected toxicity data. Billions of dollars and years of time could be saved!
…repurposed drugs are generally approved sooner (3–12 years) and at reduced (50–60%) cost (5, 6). In addition, while ~10% of new drug applications gain market approval, approximately 30% of repurposed drugs are approved, giving companies a market-driven incentive to repurpose existing assets (5)….
For example, repurposing of the emergency contraceptive, mifepristone, for Cushing’s syndrome required a cohort of less than 30 patients to test its efficacy, whereas a clinical trial1 for the same indication evaluating the safety and efficacy of a new chemical entity, levoketoconazole, required ~90 individuals (2, 3)…..
But, as it stands today, most drug repurposing efforts are done somewhat blindly; haphazardly glancing through the literature, relying on anecdotal case reports, or waiting for some academic lab to publish a five-mouse study from 2013 that hints at a secondary use. In many ways, it isn’t too dissimilar to the usual drug-discovery process! Given how promising (and relatively limited) the list of FDA-approved drugs are, the simple act of a pre-triaged list of drug-target maps (EvE’s mission!) may be extraordinarily impactful.
In such a world where this data is easily accessible, perhaps an order of magnitude more energy would be devoted to repurposing efforts, maybe vastly improving the currently horrific finances of modern day drug discovery.
But as with all seeming free-lunches, there’s a reason drug repurposing hasn’t been aggressively exploited beyond a few cases: economics. Unlike novel drugs, which come with fresh patents and a full runway of exclusivity, repurposed drugs necessarily rely on compounds whose original patents have expired or are near expiration1. This limits the sponsor’s ability to recoup development costs, because generic competition can quickly erode any profits once the drug hits the market, even if it’s approved for a new use. There are mechanisms to extend exclusivity for repurposed indications — such as the 7-year exclusivity period given by the FDA’s Orphan Drug Act for treatments of rare diseases or the 3-year-exclusivity granted in cases where new clinical data was needed to repurpose a drug — but it is a risky enough bet that most companies will shy away from it.
But as EvE is a non-profit, the economics don’t need to make sense. They plan to periodically announce opportunities for repurposing to the world, in hopes that other well-meaning non-profits take it on or, if the evidence is sufficiently convincing, that doctors simply take it as a useful datapoint for deciding whether an off-label prescription may be useful. And if they do most of the legwork in identifying good candidates for repurposing, it may even make the economics worth it for for-profit entities to pursue further.
Validation data for models
One of the easiest ways to assure yourself that what you’re doing is valuable is if people come up to you and ask if they could use whatever you’re producing. This is true in typical SaaS products, and it is true for the fruits of R&D work. But beyond assessing value outright, it also helps you learn what your work is most valuable for.
And, curiously, the primary area in which EvE has found ‘product market fit’ is in companies asking to use their data for internal model validation efforts. As I mentioned before, while EvE’s dataset is free-to-use by academics, it requires a commercial license to be used by any for-profit entity. And they are currently in discussions with 4 such commercial entities, all of whom desire to use EvE’s dataset to validate their machine-learning models predictions.
Historically, model builders in drug discovery have had to make do with whatever internal datasets they could get their hands on, which were typically limited in scope, biased toward certain classes of molecules, or simply not reproducible. Public data from sources like ChEMBL, BindingDB, or PubChem BioAssay are much larger in size, but they tend to be noisy, heterogeneous in experimental methodology, and always lack negative results. Worse, they’re often cherry-picked around success stories or clustered around well-studied targets, introducing systemic biases that hamper generalization. We need not look further than Pat Walter’s famous essay on the topic: We Need Better Benchmarks for Machine Learning in Drug Discovery, which expands on these issues even more.
This is an area of EvE’s work that I cannot personally shed much light on, and obviously, Bill cannot tell me the exact details on what the commercial entities are working on. But it was a surprising learning from our conversation that this particular topic is where public interest is most rapidly coalescing! Very excited to hear about more public statements they make in this area soon.
(Maybe) Polypharmacology
I do think this is the weakest, day-one value-add for EvE’s dataset. So take this section with a grain of salt! It just felt too interesting to not cover.
Polypharmacology is a drug discovery approach where a drug is designed to target multiple molecular targets, instead of a more traditional single-target approach. It’s not a particularly new idea, most clinically useful drugs exhibit multi-target activity whether they were designed that way or not. But what’s changed in the past decade is the intentionality.
I think there are a lot of different arguments for the value of polypharmacology, the easiest one hinging on efficacy. There’s a very interesting story that could be told here about drugs that worked better because they modulated the activity of multiple receptors in parallel. A great, recent example is that of drugs that followed Ozempic. Ozempic simply targeted GLP-1, which reduces appetite and slows digestion. But the second-generation (e.g. Zepbound) also targeted GIP, which amplifies insulin response and regulates lipid metabolism differently in adipose tissue. The effects were incredible: 13.7% weight loss with Ozempic, 20.2% weight loss with Zepbound over 48 weeks. Synergistic effects! The third generation (e.g. retatrutide) tacks on interactions with glucagon receptors — potentially increasing metabolic rate — with early phase 2 results looking once again promising.
But a more interesting place to start is the very similarly named concept of polypharmacy.
Polypharmacy refers to the clinical practice of prescribing multiple drugs simultaneously (usually 5+), typically to manage complex or co-occurring conditions. It’s common in geriatrics, psychiatry, oncology, and increasingly just about everywhere else in medicine: ~17% of all adults in the US meet the definition for polypharmacy. The logic is straightforward: most diseases aren’t governed by a single pathway, and so tackling them with a single drug is often insufficient. Instead, clinicians stack therapies: an ACE inhibitor for the blood pressure, a statin for the cholesterol, metformin for the glucose, a GLP-1 for the weight, and so on.
As you may expect, polypharmacy is awful on the patient's physiology. One study estimates that nearly 10% of hospital admissions among older adults are directly attributable to adverse drug events from polypharmacy-related side effects. The more drugs we stack onto people, the more unpredictable the net interaction becomes, because even if each one has been individually safety-tested, nobody tests all the pairwise combinations in a clinically realistic setting.
The solution may very well be to bundle things up.
Rather than throwing five separately optimized molecules at a patient and hoping for cooperative behavior, we could, in principle, design a single molecule that alone engages the same therapeutic targets. This, in turn, allows clinical trials to suss out the net effect of such a drug in a controlled, interpretable way. Which naturally leads us to the utility of polypharmacology; not necessarily because it will give us magic drugs with efficacy far better than current ones (though it may!), but rather that it will simply avoid us having to deal with the current issues that polypharmacy presents.
But the obvious question: does EvE’s dataset help with polypharmacology efforts? There isn’t any current, empirical proof of this, but I think it will. If you squint, you could see it functioning as missing infrastructure, a dataset that is necessary for rational polypharmacology to occur at scale. But this is necessarily tied up with machine-learning for chemical design accelerating, so, again, this is not necessarily something I’d expect EvE’s work to contribute to by the end of the year. But perhaps soon!
How do you understand off-target effects in a tractable way?
This all said, even if you agreed that the value proposition that EvE is claiming is real, you may struggle to verbalize exactly how you would understand the off-target effects of the 13,000~ FDA approved drugs out there. What assays would you use? How do you dose any given drug? How do you understand the translation of your assay to real-world settings?
Let’s walk through the EvE workflow.
First, you need to decide what drugs you're actually going to test. While there are technically around 13,000 FDA-approved drugs out there, many of them aren't particularly relevant for this kind of screening. You can immediately exclude things like topical medications, inhalants, radioisotopes, and simple nutrients, stuff that is known to be largely innocuous or not have much systemic impact. After this initial filtering, you end up with about 1,600 small molecule drugs that are worth investigating. But this number gets further whittled down further based on practical constraints; availability, cost, licensing requirements, etc.
From this, EvE ended up with a library of 1,397 compounds to screen.
Then comes the harder question: what exactly are you screening against? The human body has somewhere around 20,000 protein-coding genes, and there is an argument that any drug could interact with any of them. But perhaps we’d be too zealous to immediately do an (everything x everything) screen. Shouldn’t we try to do something that’s closer to the Pareto optimal frontier? What if we suspect that the vast majority of clinically meaningful drug interactions occur with a tiny subset of those 20,000 genes?
And, indeed, that turns out to be the case.
The vast majority of genes have some nominal physiological function, yes, but when it comes to drug interactions, only a minority are commonly targeted. At least a minority of classes: nuclear receptors (NRs) and 7-transmembrane receptors (also known as GPCRs). In total, there are about 800~ GPCRs and 48 NRs, but only 110 GPCR’s and 12-13 NR’s are actually targeted by drugs. Per last count, EvE has currently created data for 56 GPCR’s and 29 NR’s. Over the course of their existence, they plan to cover, in total, a select set of the 200 GPCR’s and all 48 NR’s. Why not all 800 GPCR’s? I attached that information in the footnotes.2
They hope to do much more than this too, but we’ll cover that in the last section.
Both NRs and GPCRs have some nice properties, but most pertinent to EvE, they are known to be very ‘druggable’ classes of drugs, given that the cell often uses them to convey information from the outside world, and evolution has therefore made their binding pockets unusually receptive to small molecules. GPCRs, sitting on the cell surface, are natural sensors for hormones, neurotransmitters, and other circulating ligands, many of which resemble or inspire drug scaffolds. NRs, meanwhile, act as intracellular switches that come with protected internal pockets meant to bind to estrogen, cortisol, and so on, making them ideal for selective small-molecule engagement. As a result, both are involved in a lot of important physiological processes.
This ‘physiological importance’ is useful in two ways! One, a plurality of drugs target the two — 13% of FDA-approved drugs target NR’s, with that number jumping to 35% for GPCR’s — so mapping the interactions here may give a clinically meaningful view of off-target effects. And two, given the extreme importance of GPCR’s and NR’s in modern-day drug development, there has been a fair bit of work in improving how we study their interactions with ligands of interest. As in, new assays outright shouldn’t need to be developed to study them.
Speaking of that, let’s start talking about how they are building this drug x receptor interaction map. They rely on two well-established assays which I’ll discuss here, but feel free to skip, understanding the two isn’t particularly important.
TR-FRET-based co-factor recruitment assays for NRs
When a drug successfully activates an NR, it usually causes a conformational shift that allows the receptor to recruit a specific co-factor protein, exposing what is often called the ‘AF2 domain’. These co-factors tend to have little peptide motifs (like an LXXLL motif) that latch onto that domain.
TF-FRET exploits this. A chemical is tagged onto the NR domain and a chemical is tagged onto the co-factor protein, both of which are fluorophores. If the FDA-approved drug is an agonist, you’ll see a spike of light appear as the two fluorophores interact.
Tango β-arrestin recruitment assays for 7TMs/GPCRs
Instead of recruiting co-factors inside the nucleus like NR’s, GPCR’s sit on the surface of a cell and transmit signals inward. As the name of the protein class implies, this involves utilizing G-proteins. Unfortunately, G-proteins are quite specific to their GPCR, so using them in our assay as a way to understand activation would be difficult to scale. Luckily, there is a nearly universal binding protein: β-arrestin. When a GPCR is activated by [something], their signaling process almost always involves binding to that protein.
In the assay, the GPCR (attached to a cell surface) is engineered to have a built-in “trap”, a little molecular tag connected to a transcription factor. When β-arrestin is recruited, it brings along a protease that snips the tag, releasing the transcription factor. That transcription factor then moves into the nucleus and turns on a reporter gene, which encodes for the enzyme β-lactamase. Meanwhile, the cell is loaded with CCF4-AM, a fluorescent substrate that shifts its emission profile when cleaved by β-lactamase. The stronger the GPCR activation by a drug, the more β-lactamase is produced, the more substrate is cleaved, and the bigger the fluorescence shift. That shift, measured as a ratio between ‘starting’ and ‘ending’ wavelengths, serves as a readout of how strongly the receptor was activated.
Reasonably simple! One note: the explanations I gave above is for assessing the difference between an inactive drug and an agonist. For assessing inactive versus antagonist, a separate experiment is run with a known ligand included.
Well, wait a minute. Aren’t we missing something? Off-target effects of a small molecule can be summarized purely by these GPCR/NR measurements, but we’d be failing to capture something else that is of vital importance: whether the drug outright kills the cell. One could imagine this also affecting our receptor experiments! Perhaps a drug is an antagonist and there is no color shift, or perhaps the cell is just dead, and nothing is being expressed at all. Conversely, a drug might look like an agonist due to signal drift as the cell’s internal environment falls apart.
EvE solves this in a pragmatic way: run a third assay which measures how healthy the cell is. How do you measure that? Well, one good proxy for how functional a cell is ATP production. Metabolically active cells generate ATP to power all their intracellular processes. Dead ones don’t. The assay EvE uses is called CellTiter-Glo. It works by adding a reagent that causes a fluorescent reaction in the presence of ATP. More ATP, more light. Less ATP, less light. No ATP? No light (and likely dead). Again, simple!
Is that all? One last thing: accounting for pan-assay interference compounds, or PAINS. These are molecules that often give false positives in high-throughput screening regimes. This can occur for many different reasons, but one relevant example is if a molecule itself is a fluorophore, leading to us falsely believing that it is an agonist during a run. EvE simply tracks how often a drug is leading to positive results, and flags it in their results if they believe it is a PAINS.
So they run these three assays across their pairwise (drug x receptor) combinations, producing readouts at multiple different concentrations with replicates for each one.
I’m going to skip over a lot at this point. EvE clearly put an immense amount of work into QA’ing this process and filtering through the data, and I think I would do both a disservice and detract from the point of this essay if I were to attempt to repeat it here. Summarizing it all down, using a complex logic table detailed here in Fig 7, EvE assigns 1 of 4 categories to each (drug x receptor) combination:
Inactive. Drug likely has no effect on the receptor, across all tested concentrations. Maybe it doesn’t bind or maybe it binds but does nothing.
Likely Inactive. A little more ambiguous, perhaps there’s a single noisy point above baseline, but nothing more.
Active – Unquantified. Something is happening, since there’s reproducible activity, but not enough clean data to fit a proper dose-response curve.
Active – Quantified. The drug produced a clear, dose-dependent response (as either an agonist or antagonist) with a well-behaved curve. From this, EvE fits a 4-parameter logistic model and extracts a pXC₅₀; the negative log concentration at which the drug produces half its maximal effect.
And…that’s it. A clean, rigorous, and tractable approach to understanding off-target effects, across hundreds of receptors, at multiple concentrations, using multiple modes of detection, with full transparency around the data.
How far along is EvE on their mission? Circa their last data release on 5/7/2025, 237,490 (drug x concentration x receptor) combinations have been screened, revealing 8 median agonists and 31 median antagonists per target. They run these experiments in 384 well plates, so that means they’ve run the process a little bit over 600~ times to generate their current dataset — though much of the current process is automated, very little human-done pipetting is going on. Data dumps of the data started in November 2024, with new ones dropping every few months.
I haven’t worked in a wet lab before, but I’ve been assured by at least one person I trust that the effort that went into assembling this all together is nothing short of extraordinary. But it is worth asking the question…
Why hasn’t anyone done this before?
When assessing the value of a seeming scientific achievement, it’s usually good to step back and ask one question: why wasn’t this done a decade back?
In some cases, the answer is boring: the technology wasn’t there yet to achieve it.
But here, the technology was almost certainly available! Eve’s assay for measuring NR activity has been around at least since 2008, and the one for GPCR since 2010, maybe even earlier for both. If it’s really that useful, why did it take so long for someone to start assembling this drug x receptor mapping together?
Haven’t I already given away this answer? In the introduction, I implied that pharma groups have no direct financial incentive to create such a dataset. And that is true to some degree, especially for smaller therapeutic companies that have bigger issues to focus on, but is that true for big pharma? A small slice of the billions in pharma spending couldn’t be sliced off to hand over to an internal research team? It’s not as if the data wouldn’t be useful for their own drug development pipelines. After all, off-target effects are among the most common reasons for late-stage trial failures and post-approval black box warnings, and even if the creation of an EvE-like dataset doesn’t fix the problem, I can’t imagine it’d hurt.
I should be fair: pharma companies do indeed do some of this. EvE’s own blog discusses this a little, referencing this paper:
The report’s authors, luminaries in the discipline of safety pharmacology, surveyed 18 major pharmaceutical companies regarding the numbers and identities of potential off-targets against which they test each and every one of their new drug candidates in the interest of safety. The numbers ranged from a low of 11 to a high of 104 potential off-targets routinely profiled per company, with a median of about 45. Interestingly, the industry’s opinions regarding which potential off-targets to screen vary widely. The total number of potential off-targets screened, across the universe of all 18 pharmas, was 763, yet only 12% of them were screened by more than a third of those companies.
So, yes, pharma companies do their own off-target screening. But, as we’ve discussed, this is a far cry from the universe of druggable receptors, and is only concentrated on their particular assets, not other ones. No attempt at creating a universal map!
But the same blogpost did reference another big pharma, Novartis, who also open-source a much larger map:
Novartis, who presented data collected “over a multi-year period” profiling drug/target interactions across a median of about 800 drugs per target and 105 gene product targets…
This is impressive! One may imagine that if a big pharma was willing to release this, why does an entity like EvE need to exist? For interest's sake, let’s ignore the obvious answer of ‘it is better for everyone if such a dataset is collected using a single, standardized protocol instead of compiled from unrelated experiments over years.’
I asked Bill exactly this question, and the answer was a two-parter.
For one, the dataset that was collected by Novartis, and indeed every large-scale dataset that will ever be collected by big pharma, will always be limited by the constraint we mentioned at the start: everybody only cares about the drug working. A logical conclusion of this is that nearly every receptor covered in these sorts of screens is a safety-oriented receptor. Cytochrome P450, hERG, serotonin subtypes, dopamine D₂, and the like. These are important receptors, not because of how mechanistically interesting they are, but because they are dangerous. Indeed, the vast majority of screened receptors lie within the so-called Bowes-44 set, which comes from a 2012 paper that identified 44 receptors known to be often implicated in safety-related drug failures. Though these do include NR’s and GPCR’s, it is a minimal set of them, as, again, the screening is not meant to assess how mechanistically interesting the receptors are.
And if a big pharma does decide to explore beyond the realm of safety-oriented receptors, they will almost certainly keep that dataset to themselves. Why release potential alpha to competitors? Hence, why nothing quite like EvE has come out in the past and it is unlikely it ever will in the future, at least from a for-profit entity.
And two, EvE eventually hopes to cover a lot more ground than any of the publicly available datasets. Currently, yes, the Novartis dataset is larger than EvE’s, but it won’t be for long. In fact, their plans for the upcoming few years ended up being so interesting that I decided to split it off into another section:
What does the future look like?
EvE is still quite young, just over 2 years old, and I think the future of it is going to look really, really crazy. At the end of my startup coverage articles, I typically focus on commercial/scientific risks. But given that EvE is assured funding on a multi-year horizon without needing to care about market demands, it may be much more instructive (and interesting!) to instead discuss their upcoming plans.
Earlier I noted that EvE has currently released data for 29 NRs and 56 GPCRs, out of a planned 40 NR’s and 200 GPCR’s. In my conversation with Bill, I asked him how much time is left till the remaining ones are released. I expected the answer to be, optimistically, ‘over the next few years’, given how EvE only started to release data back in November 2024 and that the Novartis dataset collection process also took several years. I was astonished to learn that he expected to have released the remainder of all GPCR + NR screens dataset by the end of this year. Setting up the assays, validation, and automation was the hard part, which is why their data releases have only started recently. But now that that’s all set up, they simply must turn the crank to get the rest out of the door.
What’s next? Bill told me that the next target of receptors are kinases, 500~ or so receptors that have been increasingly valuable drug targets over the last 20 years.
Then what? Bill said he’s open to exploring even more drug targets, but he also said, surprisingly, that EvE may add more chemicals on top of the 1,600~ planned FDA-approved drugs. The FDA-approved drugs, he said, are success stories. Potentially it’d be even more interesting to consider the failures as well. Especially the ones that everybody expected to work, arrived at phase 3, and set billions of dollars on fire after the trial results came out.
Even more exotic options are also on the table. For example, Bill discussed exploring how metabolites of approved drugs interact with targets. Some context: most secondary pharmacology work stops at the parent compound, but metabolic byproducts of a drug can have entirely different binding profiles, and, in some cases, they’re the ones responsible for efficacy (e.g codeine, which metabolizes into the much more effective morphine) or for toxicity (e.g. acetaminophen, which metabolizes into the very toxic NAPQI). He also mentioned potentially using EvE’s assaying work to develop our understanding of tool compounds, which are chemicals that don’t necessarily have therapeutic value themselves, but are used in research to probe specific biological pathways or validate target function. An ACS page has this to say about it:
While tool compounds have tremendous potential for advancing life science research, they are broadly defined, and it is often difficult for a researcher to determine the best tool compounds to employ during the research process. There remains a great need for more tool compound databases and authoritative sources of information from experts in the field.
And, as always, there is a (very short) Derek Lowe piece on how a commonly-relied upon tool compound moonlights as a ligand for a structurally unrelated receptor, likely muddying the literature the tiniest bit. More work here would almost certainly be deeply appreciated by those in the field.
Overall, EvE really exemplifies the thesis I put forward in a past essay about how smart people in biology should do more boring things. Very little that is directly sexy about doing an N x M screen, but the impact of doing something like it well can be immense. And I have little doubt that EvE Bio has been doing it well, and will continue to do so in their future projects. If you’re interested in checking out their dataset, check it out here.
Unless the owner of a still-existing patent is looking to expand indications!
In Bill’s words: (1) about half of all GPCRs are sensory receptors (taste/smell), generally regarded as not likely involved in many (or even any) diseases, and anyway smell receptors are hard to work with in HTS because their ligands are compounds with very high vapor pressures (basically, gasses); and (2) only about 170 of the remainder are validated drug targets, and only about 200 (including those 170) have compounds (either drugs or research chemicals) which are known to turn on the receptor (AKA, an agonist). It's pretty nearly impossible to design a meaningful assay for receptor activity if you don't have a positive control compound.
Abhishaike, your essays are such a delight to read. I appreciate the effort that clearly goes into putting them together. Each one is like visiting the best poster at a conference, where you come away infused with passion for a topic you’d never really thought about before. Thank you!!
Interesting and informative - thank you!