
RNA structure prediction is hard. How much does that matter?
4.8k words, 22 minute reading time


Note: I am not an expert in RNA structure, and am extremely grateful to Connor Stephens, Rishabh Anand, Ramya Rangan, and Chaitanya K. Joshi—all of whom are actual, bonafide experts—for their incredibly detailed comments on earlier drafts of this essay. All mistakes are, of course mine, and this essay should not be trusted to function as anything more than entertainment. Do your own research!
Introduction
One thing I’ve always wanted to write was ‘a primer to RNA structure modeling’. I know literally nothing about the field, other than that there are a few startups playing in the space, and have always been curious what exactly they were up to. But the release of Alphafold3—which can model RNA alongside proteins, DNA, and small molecules—dampened this desire. If a singular model solved the problem of RNA structure, who cares about the specifics of the field at large?
But while I was in San Francisco a few months back, I happened to chat with Connor Stephens, a machine learning scientist at Atomic AI. You may recognize that startup, since their founder has the distinct honor of their PhD work in RNA structure modeling being on the cover of Science in 2021 for making a substantial advance in RNA structure prediction.
But it was long unclear to me what exactly Atomic AI did in terms of R&D. This isn’t a startup post, I’m not planning to explain what their therapeutic goals are. What I was curious about was why they continue to have an ML team despite the RNA problem being seemingly solved by Alphafold3. So, I posed that question to Connor.
Connor told me something very fascinating: not only did Alphafold3 not solve the problem of RNA structure prediction, RNA may be one of the last structure prediction problems to be solved. The rest of the conversation was so incredibly fun that, midway through it, I decided it’d make for a great article to write about.
Why is RNA structure so hard to model?
On face value, the answer is pretty simple: experimentally determined RNA structures deposited in public repositories are both ridiculously small in number and of much lower quality than you’d naively expect. A quote from a paper best explains this:
There is a huge disparity in protein and RNA data. Even if there is a higher proportion of RNAs than proteins in the living, this is not reflected in the available data: only a small amount of 3D RNA structures are known. Up to June 2024, 7,759 RNA structures were deposited in the Protein Data Bank (34), compared to 216,212 protein structures. The quality and diversity of data are also different: a huge proportion of RNAs come from the same families. It implies several redundant structures that could prevent a model from being generalized to other families. In addition, a huge amount of RNA families have not yet solved structures in the PDB. This means there is no balanced and representative proportion of RNA families through the known structures.
The obvious follow-up question is: why? Apparently, RNA is a good fit for basically none of the existing structure determination methods. But again, why?
Connor told me that RNA is famous for being perhaps one of the most flexible biomolecules to exist as a category, with an almost absurd number of conformational degrees of freedom. Each nucleotide has more torsion angles than an amino acid, and the lack of a bulky side chain—like those in amino acids—means there’s very little steric hindrance to keep the backbone from flopping around. Now, keep in mind, this is not to say that RNA is unstructured. Unstructured has a particular meaning, that the energy landscape is flat, with no favored conformational structure. But this isn’t the case for RNA, which do have preferred conformational structures, there are just many of them that they constantly flip in between.
This all implies that RNA is a very bad fit for X-ray crystallography, which requires orderly, repeating conformations to arrange into a crystal. It is also a bad fit for cryo-EM (a subject I’ve written about in detail before), given both the extreme conformational heterogeneity of it and how typically small the biomolecule is, though this is increasingly being addressed. Finally, NMR, which, while more forgiving when it comes to flexibility and heterogeneity, is generally limited to very small RNA structures. Once the RNA goes beyond ~50 nucleotides, the spectra start overlapping and the resolution being insufficient to observe anything useful. And lots of important RNA lies beyond that size!
I’ve attached some nuance about NMR and cryo-EM in the footnotes.1
This means that there are really only two RNA structures that can be physically characterized: ones that have been artificially stabilized, or ones that are evolutionarily constrained to hold a single dominant conformation.
The first category includes structures coaxed into rigidity by heavy metal ions, engineered base modifications, or even crystallization chaperones. But of course, this raises a worrying question: are you really measuring the native structure, or just the structure you forced it into? The second category is rarer: RNAs that, through evolutionary pressure, have converged on a stable structure for a functional reason. There are no caveats there, only that trying to train a model on these nucleotide sequences will inevitably bias it towards unusually stable RNA structures.
Well, we shouldn’t let all of this get us down. Many impossible problems are being solved day-after-day in this field. Even if RNA modeling has all the characteristics of being hard to do—huge distributional space of possible outputs for a given input and low number of input data points—surely, some headway has been made in the problem. Consider Alphafold3: how well does it actually do on the RNA structure prediction problem?
A well-named paper titled Has AlphaFold3 achieved success for RNA? tries to answer this question. From the article:
The best models from the CASP-RNA competition, which are human-guided, outperform AlphaFold3….
….On the other hand, AlphaFold3 shows a cumulative sum of metrics greater than the other methods for the other test sets (p-value < 10−5 for RNA-Puzzles, p-value < 10−4 for RNASolo).
For RNA-Puzzles, the challenge-best solutions are from older solutions with less advanced architectures compared with the more recent CASP-RNA solutions.
For the RNA3DB_0 data set, the performance of AlphaFold3 is slightly better compared with RhoFold, which gives a better RMSD but a worse MCQ and LCS-TA.
AlphaFold3 always has a high MCQ value, indicating that it returns structures which are more physically plausible than ab initio methods (which use physics properties in their predictions).
Nonetheless, it does not always have the best RMSD (outperformed in CASP-RNA and RNA3DB_0), suggesting that AlphaFold3 does not always have the best alignment (in terms of all atoms) compared with the reference structure.
In short, while Alphafold3 is certainly an improvement in some categories of RNA—namely being the only RNA structure prediction method that can model very large RNA’s well—it does not solve the problem outright, and can be outperformed through tailored methods.
Another slightly more recent paper says something similar, and gives some insight into the practical meaning of these benchmarks, saying ‘Boltz-1 and AlphaFold3, make acceptable predictions for about half of the individual RNA chains and complexes.’. The authors further note that the results get far worse if you deviate into more structurally unique RNA space (bolding added by me):
We observed that prediction accuracy, as measured by TM-score, generally increased with higher structural similarity to the training set for all methods. The mean TM-score is below 0.1 for the category with the least similarity and increases gradually to over 0.6 for the category with the highest similarity to the training set. This suggests that AlphaFold3 and other methods tend to perform better when the target structure is more similar to motifs it encountered during training, highlighting the limitation of current methods in predicting unseen and structurally divergent RNAs.
Neat!
I could end the essay here, because this really did cover most of Connor and I’s conversation. There is a lot more that could be said about how difficult benchmarking can be in the RNA ML world, the weak co-evolutionary signal in RNA MSA’s, how even the existing set of RNA structures are made worse by the fact that they are almost always in complex with a protein, and (hearsay) that you likely need experimentally-determined templates/molecular-dynamics to get good structure predictions. This paper discusses all that in more detail if you're curious, but my main question got answered!
But the more I talked to people in the RNA space while writing this essay, the more I began to ask a new question: how important is this problem anyway?
Why even predict RNA structure in the first place?
For the protein-heads reading this, we know that protein structure actually means something quite fundamental. A protein’s three-dimensional fold is usually synonymous with its biological role: an enzyme pocket is what catalyzes a reaction, an antibody groove is what binds an antigen, a receptor domain is what recognizes a ligand. We can hem and haw about dynamics or post-translational tweaks, but the basic architecture is what makes the protein what it is. Protein structure isn’t exactly truth, but structure can be a proxy for truth a sufficiently high fraction of the time.
RNA is not like this at all. It’s actually really, really, really situational when the structure of RNA matters in a therapeutic context. Well, to be more nuanced, structure always matters, but there is a very significant split what ‘structure’ even means for this biomolecule: secondary structure and tertiary structure (image from here):
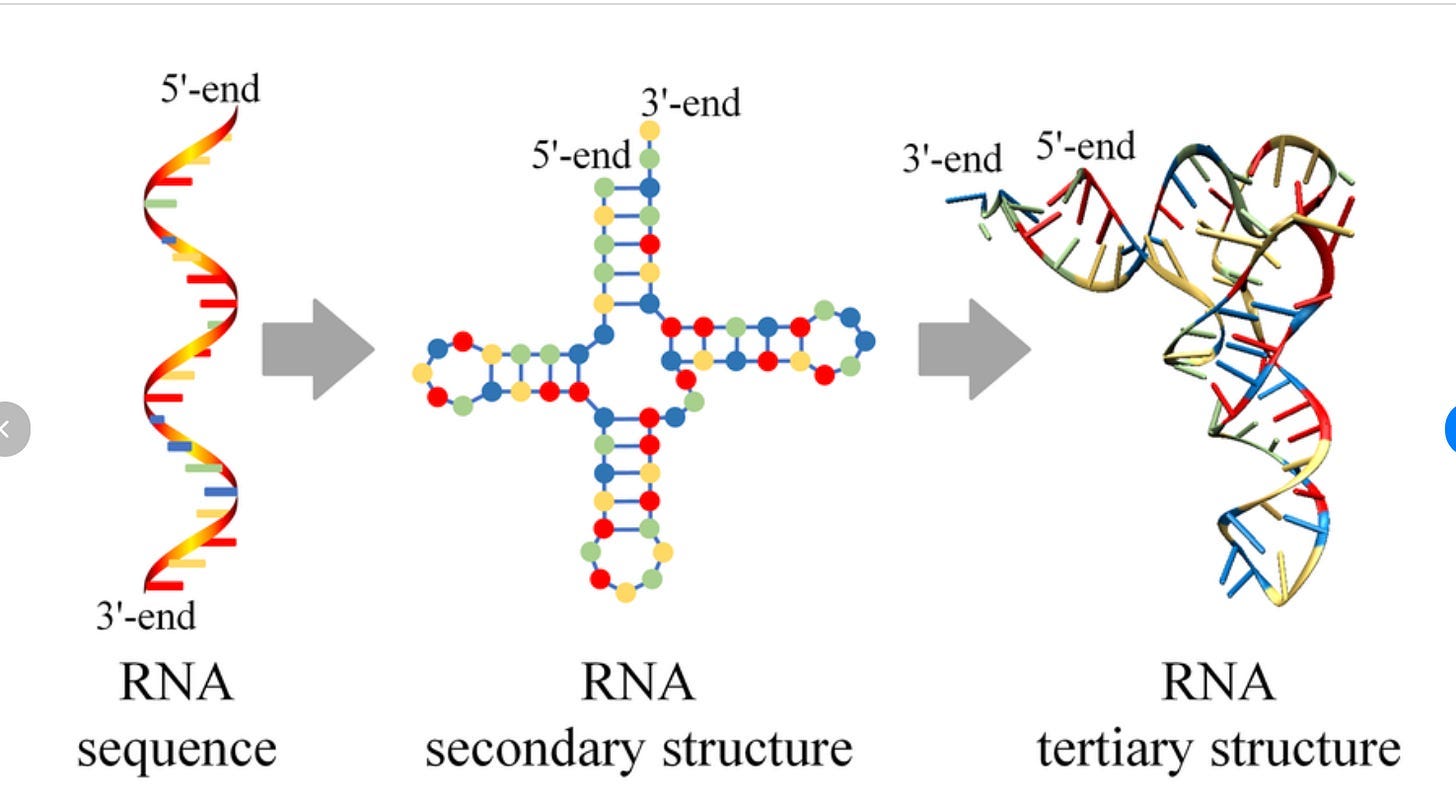
Thus far, everything we’ve talked about regarding the ‘difficulty of structure prediction’ has been for tertiary structure.
Now, this separation exists for proteins as well! But it (somewhat) matters less for proteins. Usually we treat “protein structure” as a single concept because the hierarchy is tightly coupled: secondary structure (α-helices, β-sheets) stacks neatly into tertiary folds, which in turn map directly to function. You can often ignore the distinction because the two levels reinforce each other, and so everyone hyper-focuses on tertiary structures being the most important thing.
But for RNA, the distinction matters a lot, because secondary structure seems to be where most of the clinically relevant value of structure is. Tertiary RNA structure is important! But, as far as I can tell, the value of it is actually relatively limited in scope for therapeutic-relevant problems, partially due to the fact that RNA is just so flexible that a tertiary structure phenomenon like ‘the binding site is buried in the core’ can immediately be undercut by that same core suddenly flopping out in a new conformation.
And, just as is the case for proteins, RNA secondary structure is far easier to predict than RNA tertiary structure. It’s still comparatively hard, in the sense that secondary protein structure is basically something people don’t ever worry about, and secondary RNA structure has only just recently reached those same accuracy levels. A paper that analyzed the performance of RNA models at CASP16 had this to say:
Complex and novel targets appear well beyond current capabilities for NA 3D structure prediction. However, RNA folding can be simplified into a hierarchical process: secondary structure – the pattern of canonical base pairs – forms creating a set of RNA stems which are then stitched into the overall 3D fold…
CASP16 offered the prospect of carrying out tests of secondary structure accuracy prospectively. The secondary structure of all targets, here defined as the list of all Watson-Crick-Franklin and Wobble pairs, turned out to be predicted to a high level of accuracy (Supplemental Figure 3A)...The trend in RNA secondary structure performance is more reminiscent of the performance observed in current protein 3D structure prediction, suggesting these prediction algorithms are reaching sufficient accuracy in their prediction of secondary structure to be important and useful in structural research.
Not too bad!
Returning back to our claim that ‘secondary structure is most of what you need’, let’s convince ourselves of this by walking through the major classes of RNA-based therapeutics and the importance of secondary versus tertiary structure.
The most famous form of therapy here is exogenous mRNA, and tertiary structure doesn’t seem to matter much there. I have two proof points for this. One, this mRNA optimization article from GeneWiz mentions secondary-structure optimization (e.g. preventing hairpins), but not tertiary structure. Two, just logically thinking about it, the job of mRNA is to be fed into the ribosome and translated into protein, so as long as the coding region is readable and initiation isn’t blocked (hence probably why hairpins are undesirable), why would it matter for the RNA to maintain any particular higher-order fold?
Then there’s antisense oligonucleotides, or ASO. All this is is a short synthetic strand of RNA (usually 15–25 bases long) that binds to a complementary sequence of an RNA. Once bound, it can block translation directly by preventing ribosome access, alter splicing by blocking splice sites or enhancers/silencers, or a few other things. But in all of these cases, all that matters is that the ASO can actually base-pair with its intended target. And that comes down to secondary structure accessibility: is the binding site exposed or not? Once again, this seems to be something that is largely answerable from secondary structure information, especially given how small ASO’s are.
For siRNA’s, or small interfering RNA, it’s nearly the same story as ASO’s,
Virtually the only time tertiary structure seems to matter for an RNA therapeutic is for aptamers and ribozymes. The former refers to short RNAs that fold into precise three-dimensional shapes capable of binding proteins or small molecules (e.g. theophylline aptamer), and the latter refers to enzymatic RNAs with a precise catalytic site that are able to carry out chemical reactions. But, unlike all other classes of RNA therapeutics, approved drugs here are quite rare; aptamers have two and ribozymes have zero. There’s also riboswitches, which are a hazy combination of the two, and also have no released therapies.
This all said, we should also consider the other side too: RNA as targets. How important is secondary versus tertiary structure there?
Well, things do get muddier, because there isn’t really a standardized list of established RNA targets the same way there are for proteins. There’s mRNA, the tertiary structure of which is not exploited in any FDA-approved drugs (though we’ll discuss this again later on), but what else?
Well, for one, non-coding regions! Specifically, microRNAs and lncRNA.
Given how small microRNA’s are (20~ nucleotides), I’d guess that tertiary structures don’t matter much there.
Curiously, LLM’s will, at first, insist that lncRNA’s, or “long noncoding RNAs”’ really benefit from accurate tertiary structure prediction. There’s some reason to believe that they are right. After all, they are usually above 200 (or 500, depending on who you ask) nucleotides in length, so, unlike ASOs/siRNAs/microRNAs, lncRNA’s are sufficiently large where tertiary structures may have significant impacts. Unfortunately, the LLM seems to be a bit wrong here, partially because whether lncRNA’s even form global tertiary structures at all has been a matter of intense debate for a while, though circa 2020 it is seeming like at least some lncRNA’s do. But really, whether lncRNA’s have a global structure or not wouldn’t have even mattered anyway, because their modulation does not seem to actually depend on that global structure. Rather, it depends on a set of short nucleotide motifs scattered along an otherwise floppy backbone. Even if we could perfectly predict the full structure of an lncRNA tomorrow, it feels like it wouldn’t change any therapeutic decisions. Perhaps predictions of those local 3D motifs are valuable, but that’s an open question!
As far as I can tell, the only type of RNA target where tertiary structure is known to be important is rRNA, or ribosomal RNA. Unlike most RNAs, ribosomal RNAs actually must maintain specific tertiary folds, because, like ribozymes, they are enzymes in every meaningful sense. The peptidyl transferase center of rRNA requires a highly specific three-dimensional geometry to orient its usual substrate: tRNA. And some classes of approved antibiotics, macrolides for example, are able to block this catalysis site, preventing (some) forms of bacteria from making proteins at all, eventually killing them.
It does seem like, from the outside, that accurate RNA tertiary structure predictions here would be helpful, given this line from a paper discussing where antibiotics bind to RNA:
For spectinomycin, the apparent binding site and the affected cross linking site are distant in the secondary structure but are close in tertiary structure in several recent models, indicating a localized effect. For tetracycline, the apparent binding sites are significantly separated in both the secondary and the three-dimensional structures, suggesting a more regional effect.
In other words, there is a large deviation in what secondary structure tells you, and what tertiary structure tells you!
This said, a few commenters on this essay noted that while this is an area where 3D structure is useful, it almost certainly isn’t a bottleneck due to the relative abundance of existing rRNA structures and ease of gathering new ones.
So, aptamers and rRNA are virtually the only two areas that (today) truly benefit from detailed tertiary structure modeling and have some things in the clinic. For mRNAs, ASOs, siRNAs, and most lncRNAs, the biology seems to collapse down to local accessibility and motif recognition. Both of these are sufficiently described by secondary structure, and that is decently well predicted by existing models! Tertiary folds, though definitively far from being well-predicted, don’t actually seem to influence much…at least as far as I can tell.
So why do people still work on the tertiary structure prediction problem? Is it all just for better ribosome-centric antibiotics and aptamers?
How much do we stand to gain if RNA structure prediction improves?
Well, in the immediate short term, it does seem like antibiotics and aptamers are really the field's best bets.
This is nothing to sneeze at! On the antibiotic side, we do need better antibiotics to account for the current ‘antibiotic resistance’ thing that’s been going on for the past decade, so why not elect ribosome-targeting antibiotics? This said, we should immediately drown our hopes that better ribosomal drugs will actually change the resistance trend-line. Naively, one would hope that things that interfere with rRNA functioning should be quite hard to adapt to—after all, elements of the ribosome are canonically known for being extremely conserved. And that is true, but resistance manages to evolve anyway, including via, interestingly enough, post-transcriptional-modifications that prevent the antibiotic from binding to rRNA.
Of course, the real issue with antibiotics has little to do with scientific ideas, and more to do with economics. A funny paragraph I found from an interview with the lead author of a recent ‘new rRNA antibiotic’ paper had this to say:
…there is an argument that the difficulty making successful antibiotic drugs has more to do with business models than with molecules. When asked about this, Myers says, “Do I worry about the broken business model for antibiotics development? Are you kidding? Every day. That may be the most challenging problem of the lot, and it is not one that I can solve. Synthesizing new antibiotics—in that, I feel confident.”
One related note is that RNA structure may not only be useful for targeting bacterial rRNA, but also viral RNA. A particularly famous case here is a paper that developed a protein that can bind to a structured RNA element in the HIV virus, impairing transcription of it (albeit in an in-vitro setting). Though this has yet to lead to any approved drugs, the subject is, according to one review paper, promising.
Moving onto the aptamer side, though it is still early days, the future is interesting. Circa 2024, de novo RNA aptamer design is currently at the ‘we can redesign existing things’, which is a necessary step on the way to ‘we can redesign existing things to make them better’, but we’re not there yet. What’s the therapeutic utility of an aptamer anyway? Basically the same uses one would have for an antibody for, with a ton of side benefits:
Aptamers have several advantages over antibodies, not least the fact that they can be produced quickly and easily without the need for animal use. Aptamers also benefit from low production costs, high batch-to-batch consistency, and functional stability when stored at room temperature, which gives them a long shelf-life and simplifies both transportation and storage. In addition, the low immunogenicity of aptamers makes them valuable tools for in vivo applications, while their small size compared to antibodies allows them to better penetrate cells and tissues. This can be especially useful when studying difficult-to-access targets such as those found within the tumor microenvironment.
On the flipside, aptamers are poorly suited to applications in which it is desirable to stimulate an immune response and may undergo rapid clearance in vivo unless they have been modified to prevent this.
This is quite nice, but there’s a lot of modalities vying for the antibody throne, and many of those share similar benefits as aptamers. Beyond the scope of this essay for me to judge how large the value is here, but I’m sure it’s non-zero!
Some nuance
Every essay I write, I try to form a strong opinion to build my story on, and I’ve sketched out one such opinion here: most of the value of RNA structure is in secondary structure, predicted secondary structure is quite good, and tertiary structure has a limited set of use cases. I think the argument for this position is decently strong.
But I should note that the take I have here is not a universally held opinion for those in the field, and is very much a ‘I did my research, and this is the conclusion I came to’. There are, I think, reasonable disagreements that people have had to this.
First, one paper titled Thoughts on how to think (and talk) about RNA structure argues that the seemingly high utility of secondary structure has a lot more to do with its historical ease of accessibility rather than the low utility of tertiary structure. Some context: most RNA secondary structure consists of what is called ‘Watson-Crick Pairs’, or just the tendency for RNA adenine (A) to match with Uracil (U) and Guanine (G) to pair with Cytosine (C). Non-Watson–Crick are just any hydrogen bond that forms outside of this, which typically can only be noticed in 3D space. The aforementioned paper says this about the two:
Overall, the tendency to focus on Watson–Crick pairs may stem from the fact that they are the basis of nucleic acid hybridization and that they are easier to identify, draw, and rationally mutate. However, non-Watson–Crick pairing and stacking patterns in helical junctions and internal loops preform a 3D architecture that dictates the angles of emerging helices. As a result, specific parts of the RNA are spatially positioned to readily establish interactions often involving nucleotides that are far apart in sequence, but not in three dimensions….Non-Watson–Crick pairings combined with helical stacking give rise to structural motifs that provide the building blocks of many higher-order structures, including ultrastable tetraloops and their receptors, kink-turns, E-loops, etc.
For instance, I mentioned earlier that the tertiary structure of mRNA targets is not exploited in any FDA-approved drugs. This is true, but they are being exploited in preclinical settings! For instance, Arrakis Therapeutics, a RNA-targeting-with-small molecules biotech startup with a very fun name, has this really interesting presentation showing that multiple of their ligands are able to bind to conserved, accessible 3D pockets of mRNA of the MYC protein. This is a notoriously difficult protein to directly bind to, but seemingly accessible through its mRNA.
Second and relatedly, I dismissed the value of mRNA tertiary structure, but there is an RNA modality that does something very similar to exogenous mRNA and has a very important tertiary structure: circRNA’s, or circular RNA, which form a covalently closed continuous loop. One of the giants of the field (Mihir Metkar, who was one of the primary contributors of the Moderna COVID-19 mRNA vaccine) has written a great Nature review article over mRNA broadly, and did mention that circRNA’s must rely on a fundamentally different mechanism to initiate protein translation:
Because canonical mammalian translation depends on 5′-cap recognition, mRNAs that lack a cap [e.g. circRNA’s] require an alternative means of translation initiation. One answer is an IRES (Fig. 6).
First discovered in picornaviruses, IRESs vary with respect to both their structural complexity and their reliance on endogenous initiation factors. In general, these two features are inversely correlated, with the simplest IRESs bypassing only the cap recognition step, whereas the most structurally complex bypass even AUG recognition, relying instead on intimate direct interactions with both the large and small ribosomal subunits.
In other words, the IRES’s, or internal ribosome entry site, on a circRNA is the primary way it is recruited to the ribosome. This means that translation efficiency, tissue specificity, and even coding potential can hinge on whether the IRES is stable, accessible, and folded in the right way, meaning that it is a strong axis of control of a circRNA therapeutic! For example, engineering an IRES to improve translation efficacy is something that is fully possible to do. But to do this at extreme scales, we’d likely need to be able do tertiary RNA structure prediction very well, since the three-dimensional structure of IRES seems to matter a fair bit (though, admittedly, most of the experimental structure studies of IRES are for non-therapeutically relevant ones). But why even use circRNA’s over mRNA’s? One paper explains that quite well:
Compared with the canonical linear mRNA used in vaccines, circRNAs have multiple advantages.
(1) CircRNAs are more stable and easy to store, whereas mRNA vaccines exhibit extreme instability because it is susceptible to degradation by RNases during transportation, storage, delivery, etc. Although nucleotide modifications of the mRNA backbone and UTR regions make mRNA more stable, this increases cost and complicates the manufacturing process, and the storage of the resulting vaccine still requires a low-temperature cold chain due to its suboptimal thermostability. CircRNAs without any modifications exhibit high stability and RNase resistance and can be stored at room temperature or under repeated freeze‒thaw conditions.
(2) CircRNAs without any modification exhibit fewer side effects. The cytotoxicity and side effects caused by mRNA vaccines are partly due to their high immunogenicity. Compared with modified mRNA, which has somewhat modulated high immunogenicity, circRNA exhibits lower immunogenicity, and lower cytotoxicity in the absence of modification.
(3) CircRNAs possess prolonged antigen-yielding capabilities and durable immune responses. The resulting longevity and thus prolonged antigen production contribute to antigen retention in antigen-presenting cells (APCs) and prolong antigen presentation.
Convincing to me! Very excited to see how the circRNA space plays out.
Thirdly and finally, claiming that secondary structure for RNA is nearly solved is false, at least for mRNA used in the clinic. After all, the mRNA used in vaccines is quite biochemically distinct from the mRNA we naturally produce in one important element: the uridine nucleotide is replaced with a different chemical (the most common one being 1-methyl-pseudouridine, or m1Ψ), which is more immunologically ‘quiet’. This, as you may expect, messes up secondary structure prediction a fair bit, since there are basically zero experimentally determined mRNA structures with modified nucleotides. The same Mihir Metkar paper mentioned earlier says this:
Although m1Ψ substitutions have little consequence on in vitro transcription or translational fidelity, as with other naturally occurring modified nucleotide, m1Ψ can substantially alter RNA secondary structure…these subtle differences in individual base-pair stabilities can lead to structural changes that alter mRNA functionality (for example, creating or disrupting a RNA binding protein (RBP) binding site)...
At present, the functional competence of RNA structures that contain modified nucleotides can only be assured by empirical testing.
There is ongoing work to solve this problem but the datasets are still all quite small, as is typical in the RNA world.
And that’s it! Thank you for reading!
So, one, there are some cases of cryo-EM being useful for at least some RNA structures, like here, and that may accelerate as the field of cryo-EM reconstruction gets better and better. Second, NMR can be useful for RNA structure prediction problems in cases you have a crudely-predicted structure, but think you improve it by confirming the pairwise proximity of a handful of nucleotides. This is significantly more tractable, even for larger RNA, to do via NMR!
My thesis in the Steinmetz Lab involved using computational tools to find RNA secondary structures and tertiary topologies that allow viruses to assemble around their RNA genomes. Even with the secondary structures mapped, it is difficult to determine how to assemble viruses and virus-like particles in vitro.
If you look at a phage or a virus infecting a cell, how does their capsid decide which RNAs to encapsulate? How do we not end up with empty capsids or ones containing host RNA? Secondary and tertiary structures.
I was able to isolate the minimal stem loop needed for the origin of assembly -- about 50bp -- from potato virus x and added it to a GFP mRNA. Assembly. Okay how about the nearest relative of PVX? I tried this with mycovoruses, phages, plant viruses etc. And it works as long as they are filamentous or rod-shaped viruses. I even swapped stem loops around and saw bidirectional assembly (paper almost published). I added it to circRNA and made the first halo-shaped virus-like particles that don't exist in nature. Possibilities of this are endless for therapies, agriculture, phage therapy, and so much more.
I think we have only touched on a few use cases for controlling RNA shapes - still a lot to discover
My rotation project in the Church lab was on structure prediction for bacterial T-box riboswitches (the goal was to classify which tRNA was recognized by the riboswitch; fortunately we only needed to predict the secondary structure for this goal). Even these were rather difficult to model. We looked into deep learning methods but ended up going with a HMM approach due to lack of structural data. And of course, these are ribo *switches*, which change structure upon ligand binding.
So it's very cool to see the progress that has been made since 2020.