
Creating the largest protein-protein interaction dataset in the world (A-Alpha Bio)
4.6k words, 22 minute reading time


I think this startup is promising and I hope they succeed! This is not a sponsored post, not meant to be anyone’s opinion other than my own, and is not investment advice. Here is some more information about why I write about startups at all.
Also, I wrote this post and then accidentally stumbled into a conversation with the CTO (and co-founder), Randolph Lopez! Consider almost everything in this article a view into what’s publicly available about A-Alpha Bio, and the ‘Addendum: Some notes by the CTO’ section as a view into some of the yet-to-be-released stuff.
That last section will clear up some questions/misunderstandings I got from purely the publicly available info, so make sure to read it!
Addendum: some notes by the CTO (read this!)
Introduction
Understanding how proteins interact with one another is hard. Genuinely, like stupidly hard. Way, way harder than you’d naively think it is.
One might naturally assume that because Alphafold2 largely solved the monomeric protein structure prediction problem, it let us make headway on the protein interaction problem.
After all, it’s the same underlying physics: ionic bonds, hydrogen bonds, van der Walls forces, pi-pi interactions, and so on. And it did, but the multimeric version of Alphafold2, and even Alphafold3, is still quite bad at the whole problem.
Want to know the structure of the human insulin receptor? Alphafold2 is decent at predicting it, no need to crystallize anything.
Want to know how well a 13 amino acid peptide binds to the receptor? You’ll probably need to re-run Alphafold2 with dozens of different seeds, maybe do MSA subsampling, and a laundry list of other ‘hacks’ to get these models to give a somewhat correct structure and, thus, a proxy for binding affinity. And, after all that, you’ll probably still need to do a wet-lab binding assay to confirm the inevitability noisy prediction.
A lot of this has to do with the complexity of so-called protein-protein interactions, or PPI’s. Even though the space of forces is the same, the distribution of ‘possible’ structures grows exponentially. Atom-sized deviations can cause catastrophic failures in predicted final structures, conformational flexibility can lead to unpredictable binding modes, and the sheer number of potential interaction surfaces explodes combinatorially.
Given enough data though, models should still be able to grasp the complexity. But PPI datasets are notoriously tiny. The latest collation of binding affinity dataset (PDBbind+) amounts to just 3,176 proteins in total. While there are ML models built off such datasets, they are, at best, academic curiosities, and not something essential to have in one’s protein design toolbox.
A-Alpha Bio is a biotech startup trying to solve the data problem. The company was created in 2017 by two graduate students at the University of Washington, David Younger and Randolph Lopez, the former of whom is a Baker Lab alumni. Their pitch is fundamentally a wet-lab innovation, which I’ve written about before as being a common characteristic among ML-bio companies I’m excited about.
In this essay, we’ll discuss what this innovation is, the computational angle they have, what they are using the innovation for, and the risks the company is taking.
The product
AlphaSeq
The beginnings of A-Alpha Bio stem from a 2017 paper titled ‘High-throughput characterization of protein–protein interactions by reprogramming yeast mating’. The paper introduces the first iteration of A-Alpha Bio’s method for gathering protein-protein interaction data at scale, which is also called ‘AlphaSeq’.
Let’s go over it!
How does it work?
AlphaSeq functions via exploiting yeast cell mating.
Yeast cells have two mating types: MATa and MATα. In nature, these cells find each other and fuse to form diploid cells through a process called sexual agglutination. This process is mediated by two proteins: Aga2 on MATa cells and Sag1 on MATα cells. When these proteins interact, they cause the cells to stick together, facilitating mating + the creation of a diploid cell.
AlphaSeq hijacks this natural process. Instead of using the native Aga2 and Sag1 proteins, the researchers genetically engineered yeast cells to display proteins of interest on their surface. This alone isn’t particularly novel, this has been done for decades underneath the name ‘yeast surface display’. The main novelty here is in exploiting the mating aspect of it: MATa cells display one set of proteins, while MATα cells display another set. When you mix these engineered cells, the likelihood of them mating becomes a function of how strongly the displayed proteins interact.
But how do you measure these interactions across thousands or millions of protein pairs? The answer is, of course, DNA-encoded libraries. Each protein displayed on the yeast surface is associated with a unique DNA barcode. When two yeast cells mate, these barcodes are brought together in the resulting diploid cell. Through some genetic engineering, these barcodes end up next to each other on the same chromosome.
From there on, you can simply extract the DNA from all these mated cells and sequence it. The frequency with which you see two barcodes together directly correlates with the strength of interaction between the proteins those barcodes represent. More frequent pairings indicate stronger interactions.
Thus, we can get an N by M protein interaction screen, where N MATa cells are each displaying 1 of N proteins, and so on for the M MATα cells.
When I first read about this, I had a list of questions. I’ll go through them, one by one.
That's a weird mating pattern. Why do yeast cells do that? It’s not that weird, it’s not too dissimilar to male and female sexes in mammals, sex recognition here just occurs via cell surface proteins. And why ‘types’ exist for yeast cells is for the reason sexes exist for animals: a way to nudge life towards greater genetic diversity. One note: this isn’t actually equivalent to sex, I used the phrase ‘mating types’ for a reason. Different sexes imply differently sized gametes (large eggs, small sperm), but the gametes between yeast mating cell types are the same, only surface proteins are different.
Does each given MATa and MATα cell only express one protein at a time? Yup! The paper doesn’t explore multiple surface protein expression, but, just thinking about it naively, it sounds like it’d be annoying to make something out of the resulting dataset.
Can this method account for post-translational modifications? The original paper is pessimistic on this front, writing ‘the detection of interactions requiring specific post-translational modifications may not be possible’. On a more theoretical level, it is known that yeast cells are incapable of recapitulating certain post-translational modifications that mammalian cells can do (e.g high-mannose glycans), so I’d expect AlphaSeq to have a similar failure mode.
What is the actual output of an AlphaSeq run? It’s a value referred to as the ‘mating efficiency’ of the yeast cells. More specifically, it is (number of diploid cells) divided by (number of MATa cells + number of MATα cells + Number of Diploid Cells), for a given protein-protein pair. So, you’d end up with one of these values for every protein-protein pair.
Is it possible to connect mating efficiency to a traditional binding affinity value? The researchers established a calibration curve using protein pairs with known binding affinities. They found a log-linear relationship between mating efficiency and experimentally-determined binding affinity (Kd) with a pretty high correlation (R2 of .87). From the paper’s analyses, there seems to be a wide dynamic range here; the relationship held over multiple orders of magnitude, from 500 pM (strong) to 300 μM (weak). This means that the output of AlphaSeq the mating efficiency values can directly represent a true affinity value, ranging from very strong to very weak.
What scale can this operate at? The original paper could generate 7,000 distinct PPI affinity values in a single experimental run. Which is quite large, it immediately outstrips the existing set of publicly available data. But it’s also relatively small given the dimensionality at which biology operates at. A 2023 paper by A-Alpha Bio pushed this further, generating binding affinity data for 104k~ antibodies to a single SARS-CoV-2 peptide in a (seemingly?) single run! Finally, A-Alpha Bio’s most recent work claimed to run 15,000 antibodies by 200 target runs, so 3 million interactions in total. Curiously, this was a throwaway example in the attached poster and not the actual discussed result. Which is…weird. The actual experiment they ran was far smaller in scale, with less than 500~ total interactions.
Overall, cool method! We’ll discuss some outstanding concerns in a bit, but let’s first ponder on what this method is trying to replace.
What are the alternatives?
There are a few competitors in the ‘high-throughput in-vivo PPI screening’ world.
It’s a surprisingly diverse area, but two main names seem to consistently pop up, both of which also rely on yeast: yeast two-hybrid (Y2H) and yeast display. We could mention phage display as well, but it has the same practical constraints as yeast display, so I’ll leave out explicitly discussing it for now.
First, Y2H. In this method, there’s a ‘bait’ protein and a ‘prey’ protein, both of which are proteins you’re interested in assessing interactions between. The "bait" protein is fused to a DNA-binding domain (BD) of a transcription factor, and the "prey" protein is fused to an activation domain (AD) of the same transcription factor.
The DB protein, as the name implies, immediately grabs onto DNA. If the bait and prey proteins interact, the BD and AD are brought together, activating the transcription of a reporter gene, which can be detected with other methods. If they don’t interact, the AD will be (theoretically) incapable of ‘finding’ the reporter gene. As the DB and AD fusion proteins are expressed intracellularly, Y2H occurs entirely in the nucleus.

You may immediately notice that Y2H doesn’t incorporate barcodes, so the scalability of the whole method is quite low; the possible transcription of the reporter gene must be found one at a time. There is a method called BFG‐Y2H that fixes this, incorporating barcodes into the whole system and allowing us to rely on high-throughput sequencing. This method claims a scale of 2.5M protein-protein pairs, which is quite high!
However, Y2H as a method suffers from relatively low accuracy, something along the lines of a 50% true-positive rate, an accuracy that BFG‐Y2H most likely shares. This is primarily due to intracellular environments being a bit uncontrollable, a cell is a crowded place after all, and spurious interactions are the norm. AlphaSeq likely doesn’t suffer from this problem, given that binding occurs in a less dense + more controllable extracellular region.
Moving on, yeast display is our next contender. The general idea here is simpler than Y2H. Transfect a cell with your desired gene, the protein displays on the surface of the cell, wash the cell with a purified protein you’re interested in, and then use flow cytometry (or something else) to detect binding. The cell surface has one protein of interest, the wash has another protein of interest, and flow cytometry reveals whether binding occurred (via an attached chromophore on the washed purified protein).
Yeast display can operate at the 10M+ scale of interactions and, as the interactions are occurring extracellularly, won’t have the same accuracy drawbacks as Y2H has. The disadvantage with this method seems to primarily boil down to two things: dependence on purified proteins and spectral resolution constraints.
For the former, purified proteins are expensive, may be structurally unstable, and some classes of proteins cannot be purified at all (e.g. membrane proteins). It’d be ideal if everything could simply be expressed within the cell itself, as is done with AlphaSeq.
For the latter, flow cytometry has a finite number of distinct fluorescent channels that can be used simultaneously. Use too many channels and you’re drowned in noise! This means that only a limited number of targets can be used per round, typically around 15-20. While more powerful versions of flow cytometry can push that number up more, it still isn’t high! Yeast display is fundamentally incompatible with doing something like a 1000 nanobody to 1000 receptors screen, more like a 1000 nanobody to 10 receptor screen. Comparatively, as AlphaSeq relies on DNA encoding to represent binding, the number of targets can be far higher.
So, AlphaSeq does seem better in some respects. But what about where it’s worse than other methods?
Outstanding concerns
Obviously, there are problems with yeast as a platform at all. Specifically, in that it cannot recapitulate the same post-translational modifications as done in human cells, which may affect the folding of an expressed protein, which may affect binding.
But, given that the main competitors are also based on yeast and will suffer from the same problems, let’s ignore those.
What’s specifically uniquely bad about AlphaSeq?
Truthfully, I struggle to come up with anything concrete.
One complaint is potentially that AlphaSeq is worse at scaling measurements — if we are to take existing papers as proof of how far it could be pushed — than other yeast-based PPI detection methods. I imagine that are some inherent physical limitations based on a yeast cell’s probability of being able to interact with every other yeast cell of the opposing mating type.
To be clear, maybe it is on par, capable of reaching 108 measurements and beyond. The papers thus far haven’t revealed much…we know 106 scale is absolutely possible, and maybe 107. The 107 case was a throwaway example and not something they expanded upon heavily, so I’m currently taking it with a grain of salt. Perhaps the error rate of that level of scale is extremely high?
There’s also this vague worry I have about the mating efficiency → binding affinity calculation. It’s surprising to me that it works at all! Variations in how well different proteins are expressed on the yeast surface and the experimental medium of the cell could change folding patterns of proteins, so I wonder how widely applicable the method is in practice amongst diverse proteins.
AlphaBind
AlphaBind doesn’t seem to be a single thing, but seems to broadly refer to a suite of ML models that A-Alpha Bio has built on top of their binding affinity data.
There is…basically zero information online about this. They claim to have ‘750 million affinity measurements’ upon which these models are trained, which is immense, but there aren’t any concrete results showing how useful the resulting model is.
Hoping for a paper in this area to be released soon!
How do they make money?
They seem to be placing bets on both the therapeutic angle and the platform angle.
For therapeutics, it’s immunocytokines.
For platforms (in partnerships with Big Pharma), it’s molecular glues.
At least, that seems to be their focus based on the pipeline on their website.
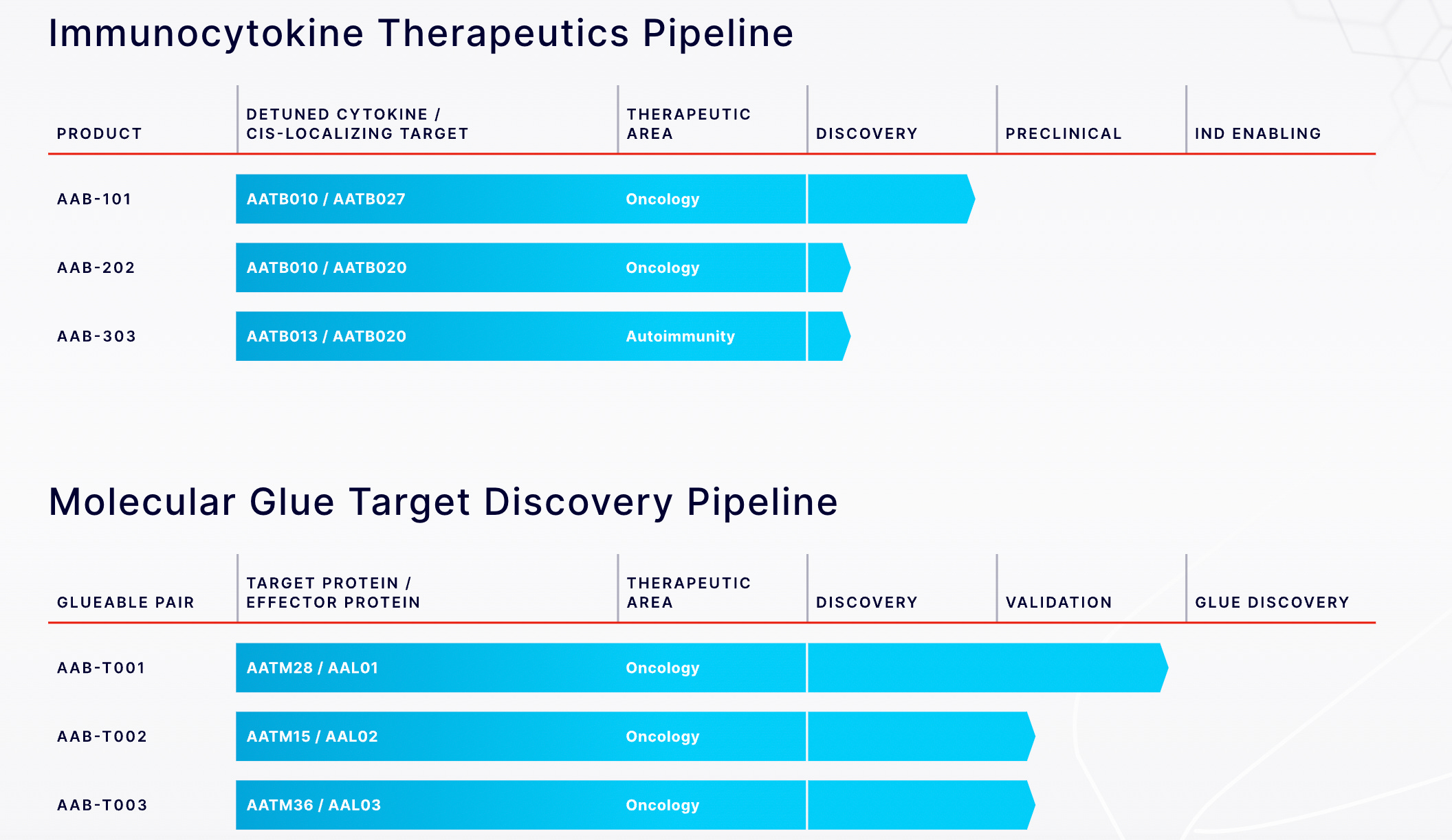
Why those two? Likely because both of them (seem to) benefit from high N by M interaction screens, where both N and M are quite high. This is opposed to, say, few-target optimization, where you might only need to screen a large library against one or a handful of targets.
Let’s go through a quick explanation of both modalities + their work in it.
Immunocytokines
Immunocytokines are fusion proteins that combine an antibody with a cytokine (a protein that interacts with the immune system). The antibody component provides targeted delivery to specific cells or tissues, while the cytokine portion delivers an immunomodulatory signal to nearby immune cells. This combination aims to concentrate the cytokine's effects at the desired site of action (such as a tumor), potentially enhancing efficacy while reducing systemic side effects.
For example, consider an antibody tuned to bind to antigens on a tumor.
If we fuse this antibody with IL-2, a cytokine, the antibody will grab onto the tumor and any passing T or NK cells will bind to the free-floating IL-2.

What happens when IL-2 binds to these passing-by immune cells? A lot of things. The immune cells ramp up its ability to be cytotoxic, it replicates, and produce even more cytokines to alert other immune cells. The hope is through this local activation of the immune cells, the brunt of their ‘response’ is directed towards the tumor.
Unfortunately, the efficacy of immunocytokines has been limited by systemic toxicity. This is because the attached cytokine can bind to receptors throughout the body, not just the immune cells. In other words, cross-reactivity is a big problem.
The utility of the AlphaSeq platform comes into play here! If you want to build cytokines that can activate a specific target of interest (e.g. immune cell receptors) while avoiding everything else in the body, a scalable protein-protein interaction screen would come in handy. Engineering thousands of cytokines and assessing their affinity across thousands of receptors, trying to increase affinity to one of these receptors and diminish it on everything else.
A-Alpha Bio has poked at one part of this problem in a recent poster titled ‘Cytokine affinity tuning using the AlphaSeq platform to generate targeted immuno-oncology therapeutics’, performing single-site mutagenesis on a wild-type cytokine to assess each of their affinity to two receptors at once.
It works according to their results, but…it’s also somewhat unimpressive.
This was essentially a 500~ cytokine (at the upper end) by 2 receptors PPI screen. Which is a fine problem set-up, but also a bit disappointing if scale is really the selling factor here. I intuitively get the sense that AlphaSeq is a fair bit more accurate than Y2H, given how terrible Y2H is, and easier to set up than Yeast Display, since purified protein isn’t needed, so that’s a win in of itself!
But if scale is what is being advertised, the existing paper on the topic doesn’t quite prove it. My guess is that the paper is more of a proof of concept than anything else, given that this one came after their much larger, 100k~ antibody AlphaSeq screen. Looking forwards to more work in the future!
Molecular glues
Molecular glues are a class of small molecules that facilitate protein-protein interactions (PPIs) that wouldn't occur naturally or would occur only weakly. Unlike traditional drugs that inhibit protein function, molecular glues work by bringing two proteins together, leading to the degradation of a target protein.
The classic example of a molecular glue is thalidomide. Yes, the drug that causes horrific birth defects was one of the first molecular glues ever used in clinical practice, though its mechanism of action wasn’t known at the time.
Quick explanation on how it works: Thalidomide allows the cereblon protein (CRBN), an E3 ligase, to bind to IKAROS proteins, a set of transcription factors. Under ordinary circumstances, these two proteins do not interact. But thalidomide allows it occur by acting, as its name implies, a glue!
Why is the interaction useful? Through some complex chemical magic facilitated by the attached E3 ligase, multiple ubiquitin proteins are attached to the IKAROSE protein, also known as ubiquitination. This particular protein tag is recognized by the proteasome, which are protein complexes that degrade other proteins into individual amino acids or peptides. Thus, thalidomide reduces levels of intracellular IKAROS proteins by tagging it for degradation.
To note, this is why CRBN being an E3 ligase is important, since E3 ligases — of which there are 600~ — are capable of ubiquitination of attached proteins.
Why reduce IKAROS protein levels? Well, cancer often have overactive transcription protein levels due to their rapid growth, so reducing IKAROS levels helps stem their proliferation. But the initial prescription of thalidomide was for a more minor and widespread condition: pregnancy morning sickness. Of course, as it turned out, IKAROS proteins are also responsible for increased levels of the FGF8 protein, which is essential for correct limb and brain organization during embryonic development. Thus, the tragedy of thalidomide babies.
Despite the past of thalidomide, the concept of a molecular glue remained curious, given its advantages over typical inhibition-based drugs, and research continues to this day, still primarily focused on gluing stuff to E3 ligases.
On face value, the role of A-Alpha Bio’s AlphaSeq here is a bit uncertain. Molecular glues are small molecules, why is a PPI company interested in them? Curiously, they are placing themselves as not the people discovering glues, but rather the people discovering pairs of proteins where glues can be applied in the first place.
The throughput and sensitivity of AlphaSeq makes the platform well suited to uncover novel targets for molecular glues by discovering and characterizing weak interactions between E3 ubiquitin ligases (or other effector proteins) and target proteins that may be enhanced by a small molecule binding at the interface. We measure interactions between our proprietary E3 ligase library and undruggable target proteins to provide a starting point for the rational discovery of molecular glues. We have discovered and validated many pairs internally and look to partner with industry leaders to progress protein-protein binding insights into small molecule drugs.
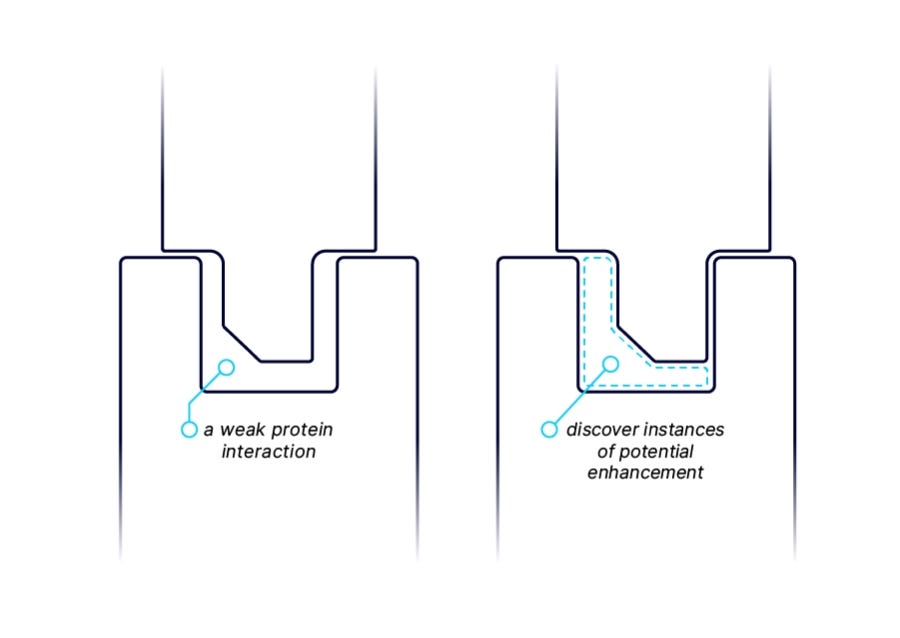
I was initially a little skeptical of the value of this.
Intuitively, my understanding of molecular glues is that that weak interactions aren’t strictly necessary; they can cause interactions between anything. What’s the purpose of learning about weak interactions?
As it turns out, while molecular glues can induce interactions between non-interacting proteins, the primary way they function is by stabilizing weak interactions!
One review paper over molecular glues explicitly states:
Many molecular glues take advantage of weak, fortuitous, pre-existing protein–protein interfaces (PPIs) that can be further strengthened by their binding
Discovering weak protein–protein interactions that can be further stabilized is key to develop molecular glues. Rational design of molecular glues has been difficult mainly due to a lack of understanding and predictability of weak interactions.
Another review paper discussing the challenges of molecular glue development says something similar:
We believe that utilizing methods such as high-throughput global proteomics to understand the target interactome ahead of establishing screening will be more successful. This will undoubtedly require assays that are able to measure weak and transient interactions between target and effector.
This also answers a side piece of skepticism I had about whether pairs are actually the important piece (versus the molecular glue development itself)! It seems that the molecular glue field is so early that good targets to develop glues on top of are, for the most part, still unknown.
This is where A-Alpha Bio comes in, as they are the only group who can screen many E3 ligases against many target proteins. They aren’t developing a therapeutic themselves, but helping form the basis for a therapeutic at all! This is likely why many of their existing partnerships — Amgen, Squibb, and Kymera Therapeutics — all focus on molecular glues.
It’s hard to assess how well this is going, since there are no publications focused on it, but partnerships usually mean there is something here!
What bets is the company making?
A-Alpha Bio feels deeply interesting to me. Again, it’s built off a fundamental advance in wet lab innovation, which I’ve discussed previously as being very important, and has lots of ongoing partnerships. Their future feels promising!
But all companies, by virtue of existing at all, are implicitly making bets on where the future is heading. A-Alpha Bio is no different. Let’s end this essay by going through them!
Here are the bets I’m seeing. These are in no particular order! Generally, there aren’t any huge concerns I have.
AlphaSeq will be able to scale further. There’s a deep disconnect between the claimed capabilities of AlphaSeq (millions of interactions per experiment) and the published capabilities (100k~ in one run). While there is this constantly mentioned line about being able to scale up to 3M interactions, there is no published paper about that experiment specifically! Very curious to know why! It may be the case that such experiments are simply too expensive to run for an academic paper and that AlphaSeq is perfectly capable of scaling as much as desired. And even if 3M can be reached, could it be pushed even further?
N by M interactions will continue to be valuable, compared to N by <20 interactions. AlphaSeq’s primary alpha lies in its ability to do many-by-many screening. This isn’t naively always valuable; many drug programs focus explicitly on a few targets and optimize molecules for those specifically. For those types of screens, you could do typical yeast/phage display. AlphaSeq’s sort of exploratory screens are well suited for A-Alpha Bio’s programs (immunocytokines and molecular glues), so this is a bet that those programs either yield results or that there are other areas where N by M screens are useful.
There is high translatability between yeast-cell expressed proteins and in-vivo human proteins. It's still unclear how well the data collected by A-Alpha Bio translates to human biology, given the differences in post-translational modifications between yeast and human cells. While this limitation will be present in any yeast screening methods, it becomes especially pertinent if your entire company is built off a yeast-based method! Still, low priority in general given that they have done some validation of translatability for a few proteins.
AlphaBind models works better than other people’s models. The lack of public information or academic publications about the suite of AlphaBind models is somewhat concerning. It may be the case that more foundation-model-esque things like AlphaFold3 are strongly outperforming internal AlphaBind results, or even simple dataset cleanups of existing open-sourced data, like PINDER, get you as far as AlphaBind. Alternatively, A-Alpha Bio may just be keeping things under wraps! Hard to tell.
That’s about it! Overall, this is a company I’m enormously bullish on, and I have high hopes for their future success.
Thank you for reading!
Addendum: some notes by the CTO
This is a section I made last minute!
After teasing the post on Twitter the other day, the CTO/co-founder of A-Alpha Bio, Randolph Lopez, followed me, and I took a chance to ask a few questions that were on my mind after writing the post.
I’ll organize this as an FAQ, with a ‘C’ for comments.
Q: What is the scale that AlphaSeq is capable of?
A: We currently execute about 30 AlphaSeq assays per month ranging for 100k intersections to about 5M per assay. The larger the network size, the more difficult it becomes to detect weak interactions so different applications require different network sizes.
C: This answers the 3M question; it’s not just a throwaway example, it’s real and possible! The ‘30 AlphaSeq assays per month’ comment also feels insane given the scale of each output. That’s 30M-150M datapoints each month!
Q: What’s the limiting factor to pushing AlphaSeq further?
A: Fundamentally, the two constraints to network size are assay volume, which determines yeast collision sampling and sequencing coverage.
C: The yeast collision sampling part sounds similar to what I mentioned earlier: ‘inherent physical limitations based on a yeast cell’s probability of being able to interact with every other yeast cell of the opposing mating type.’. But the sequencing coverage part wasn’t something I would’ve guessed is a bottleneck! It makes sense though, number of reads per unique pair will dramatically fall as the matrix grows and the confidence in any given result likely plummets.
Q: Is there a limitation on the diversity of proteins used in the AlphaSeq screen? Like, is 1000 random proteins by 1000 random proteins viable?
A: It is viable, main challenge is DNA synthesis cost. We’ve been experiment with oligo batch assembly approaches to build larger protein libraries from oligo pools to circumvent this.
C: No real comments, cool! My original concern about diversity (‘I wonder how widely applicable the method is in practice amongst diverse proteins.‘) seems to be a non-issue.
Q: Will anything about AlphaBind will be published soon?
A: That’s the plan! Aiming to have something out before EOY.
C: Hype!
I have found this blog post extremely interesting, I simply LOVE IT!
I am someone that has read tens of thousands of papers on pubmed and I am generally extremely disappointed with substack and the internet in general.
I find myself completely information starved, blog posts very rarely teach me something in science because blogs actually extremely rarely talk about science and science papers.
Most are limited to surface level topics with no depth or to regurgitating the same talking points ad nauseam (e.g. the mental black holes of exercise and diet for longevity...)
So overall internet to me is a very lonely experience and it's breath of fresh air to find someone that actually teach me important and in depth scientific knowledge. If btw you have substack/authors to recommend to me I am all ears.
About the post:
I was wondering about how AlphaSeq DB compares in terms of protein protein interactions versus the most cited one: STRING
https://pubmed.ncbi.nlm.nih.gov/36370105/
More generally we are seeing considerable advances in AI and metrological scalability in biotech and that is something you could write a lot about, I'll give you suggestions another time
It would be interesting to know how many of the comments you make in the introduction also apply to Alphafold3.